© 2022 Janghyun Choi
Counting Reads in Features with htseq-count
HTSeq is a Python library designed to process and analyze data from high-throughput sequencing (HTS) experiments. This tool counts the number of reads mapping to each genomic feature. It requires input files, such as BAM files sorted by name or position, to be provided. To ensure comprehensive analysis and avoid zero-frequency issues, it is crucial to include all relevant input files (e.g., control, treat A, treat B, … , treat N) consecutively. While this operation did not require high CPU usage, demands higher memory (> 16 Gb) to fast count the reads (i.e. this step takes a very long times, approx. 6 hrs on 12 samples). This protocol was created based on HTseq version 0.11.1 running on a system equipped with an Intel 10th generation i9-10910 processor and 48GB of memory. The test environment includes Python version 3.8.5, SciPy version 1.6.2, NumPy version 1.20.1, and pySam version 0.16.0.1 under macOS 12.4 environment.
Installation HTSeq
![]()

To install HTSeq via anaconda or PyPI, use the following commands:
$ conda install -c bioconda htseq
# OR
$ pip install HTSeq
Obtain a Gene Annotated File from Ensembl
- Select a desire species in the section of All genome from Ensemble website.
- Click “Download GTF (or GTF3)” in the Gene annotation section.
- An FTP server appears where you can download the GTF files.
- The GTF files that can be used for RNA-seq analysis have the following names: [species].[species version].[release version].gtf.gz. (e.g. human: Homo_sapiens.GRCh38.111.gtf.gz, mouse: Mus_musculus.GRCm39.111.gtf.gz, zebrafish: Danio_rerio.GRCz11.111.gtf.gz)
- Download and uncompress (optional) it and move desired folder.
Users may find it more convenient to download GTF files for human and mouse transcriptome analysis from “gencode”.
Running htseq-count
Use the following command to perform counting transcripts in mapped genome with htseq-count:
$ htseq-count -f <format> --strand=<yes/no> --mode=<mode> -r <name/pos> \
<input_1.bam>...<input_N> <AnnotateFile.gtf> > <output.txt>
In these commands,
| Parameter | Description |
|---|---|
-f <format> | Specifies format of the input data. Possible values are SAM or BAM. |
--strand=<strand> | Specifies whether the data is from a strans-specific assay. Possible value for <strand>are yes, no, and reverse. (default: yes). |
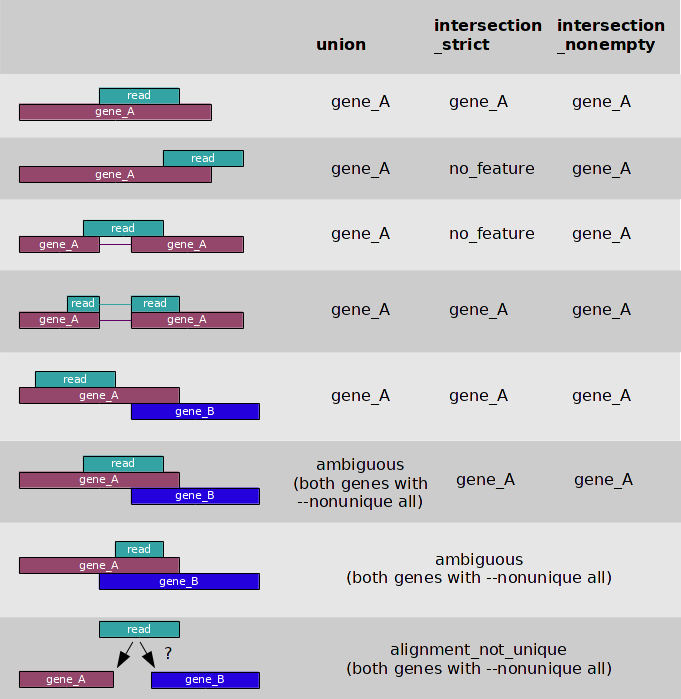
--mode=<mode> | Mode to handle reads overlapping more than one feature. Possible values for <mode> are union, intersection-strict and intersection-nonempty (default: union). Several researchers most used as intersection-nonempty. Overlapping methods refer as bellow table. |
-r <order> | Specifies how the BAM files are sorted. The BAM files used in this operation must be sorted either by name (name) or position (pos). As ‘name’ option expects all the alignments to appear in adjacent records in input, most users use name. |
<input_1.bam>...<input_N> | Specifies input data, it is separated by spaces. |
<AnnotateFile.gtf> | For count the reads, must be required gene annotation files as a GTF format. |
> <output.txt> | Specifies output file, this file generates as a plain text file. > symbol must appear before the <output.txt> syntax. |
Overlapping Table
The following table illustrates the effect of these three modes (union, intersection-strict and intersection-nonempty) :
Example Code
Here is an example command to perform htseq-count with the zebrafish genome on 4 samples:
$ htseq-count -f bam --strand=no --mode=intersection-nonempty -r name \
sort_MA_rep1.bam sort_MA_rep2.bam sort_Y_rep1.bam sort_Y_rep2.bam \
/Users/jchoi/Desktop/Danio_rerio.GRCz11.111.gtf > /Users/jchoi/Desktop/Age.txt
- When htseq-count is running, it will print out the progress in real time. The output is very long, so I will not describe it.
Citation
- Anders, S., Pyl, P. T., & Huber, W. (2015). HTSeq—a Python framework to work with high-throughput sequencing data. bioinformatics, 31(2), 166-169. DOI