© 2022 Janghyun Choi
Advanced RNA-seq Data Analysis with EdgeR: Identifying Differentially Expressed Genes and Beyond
EdgeR is a powerful tool designed for the data analysis phase of RNA-seq experiments, specifically for identifying differentially expressed genes (DEGs) across various conditions. It employs a negative binomial model to account for overdispersion in count data and uses empirical Bayes methods to stabilize variance estimates. Key features include effective normalization techniques, robust statistical testing for DEGs, and comprehensive visualization tools. While both EdgeR and DESeq are popular tools for RNA-seq data analysis, EdgeR is particularly noted for its flexibility and performance with small sample sizes due to its advanced empirical Bayes methods for dispersion estimation. This makes EdgeR especially useful in experiments where the number of biological replicates is limited. Additionally, EdgeR offers extensive options for data exploration and visualization, enhancing its utility in deriving meaningful biological insights. This protocol was created based on edgeR version 4.1.2 running on a system equipped with an Intel 10th generation i9-10910 processor and 48GB of memory. The test environment includes R version 4.4.0 under macOS 12.4 environment.
The Importance of Normalization
RNA-seq data analysis requires normalization to fairly compare samples by accounting for differences in sequencing depth and library size. For example, suppose the control group and drug-treated group have the following counts for genes A, B, C, D, and E. In the control group, the counts are 100, 200, 300, 400, and 500, respectively. In the drug-treated group, the counts are 100, 200, 300, 400, and 2000, respectively. The total counts for the control group and drug-treated group are 1500 (100+200+300+400+500) and 3000 (100+200+300+400+2000), respectively. At first glance, the gene expression levels seem identical between the groups. However, normalization reveals a different picture (see the bellow table).
Normalized Values
| Gene | Control Group | Drug-Treated Group |
|---|---|---|
| A | 100/1500 = 0.067 | 100/3000 = 0.033 |
| B | 200/1500 = 0.133 | 200/3000 = 0.067 |
| C | 300/1500 = 0.2 | 300/3000 = 0.1 |
| D | 400/1500 = 0.267 | 400/3000 = 0.133 |
| E | 500/1500 = 0.333 | 2000/3000 = 0.667 |
- Normalized values indicate that genes A, B, C, and D are relatively less expressed in the drug-treated group, while gene E is significantly more expressed. Although the raw counts are the same, normalization considers the total count differences between samples.
- Sometimes people try to determine gene expression trends by looking at read counts alone. This is never a scientific method and always requires normalization and statistical analysis.
In practice, EdgeR employs more sophisticated normalization methods beyond simple division by total counts. EdgeR uses the TMM (Trimmed Mean of M-values) method to calculate normalization factors, adjusting for sequencing depth and library size differences:
Calculate \( M \) and \( A \) values:
\[ M = \log_2 \left(\frac{X}{\bar{X}}\right) - \log_2 \left(\frac{Y}{\bar{Y}}\right) \]
\[ A = \frac{1}{2} \left(\log_2(X) + \log_2(Y)\right) \]
\( X \): Read count for a particular gene in sample 1, \(\bar{X}\): Geometric mean of read counts in sample 1
\( Y \): Read count for the same gene in sample 2, \(\bar{Y}\): Geometric mean of read counts in sample 2
Compute the weight (\( w \)):
\[ w = \frac{\text{Trimmed mean of } M}{\log_2(\bar{Y}/\bar{X})} \]
The weight \( w \) is used to normalize the read counts, reducing technical variability and allowing for more accurate comparison of gene expression levels between samples.
This complex normalization reduces technical variability and enhances the detection of genuine biological differences. EdgeR combines this advanced normalization with robust statistical techniques to accurately identify differentially expressed genes.
Installation edgeR
![]()
- Check and install edgeR Start by ensuring the BiocManager package is installed, then install edgeR using it:
if (!require("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install("edgeR") - Install Required Libraries Install additional libraries necessary for edgeR to function properly. You can install these packages manually using the
BiocManager::install("PackageName"). The list of packages is shown below.
Library list (PackageName)
-
limma: EdgeR dependent package -
HTSFilter: A filtering tool for a global Jaccard similarity index -
MASS: Functions and datasets to support Venables and Ripley -
lattice: For trellis graphics system -
mixOmics: For multivariate methods -
biomaRt: For annotation -
ggplot2 -
RColorBrewer -
rtracklayer -
GenomeInfoDb
Step-by-Step Running Guide edgeR
1. Load edgeR and its dependency packages
library(edgeR)
library(limma)
library(biomaRt)
library(ggplot2)
library(RColorBrewer)
library(stats) # For PCA plot
library(data.table)
2. Prepare metadata
- Metadata in RNA-seq is used to organize samples, adjust statistical models, ensure reproducibility, facilitate comparisons, and manage data effectively.
the metadata must be associated with the raw count table. This file can be created in Excel, saved as a csv and txt, and imported via read.csv and read.table, or it can be created by typing directly in R as shown below.
File <- c("shControl_Unt_rep1", "shControl_Unt_rep2", "shControl_H2O2_rep1", "shControl_H2O2_rep2",
"shMLL1_Unt_rep1", "shMLL1_Unt_rep2", "shMLL1_H2O2_rep1", "shMLL1_H2O2_rep2", "shUTX_Unt_rep1",
"shUTX_Unt_rep2", "shUTX_H2O2_rep1", "shUTX_H2O2_rep2", "shNRF2_Unt_rep1", "shNRF2_Unt_rep2",
"shNRF2_H2O2_rep1", "shNRF2_H2O2_rep2")
Rep <- c("rep1", "rep2", "rep1", "rep2", "rep1", "rep2", "rep1", "rep2", "rep1", "rep2", "rep1", "rep2",
"rep1", "rep2", "rep1", "rep2")
Condition <- c("Unt", "Unt", "H2O2", "H2O2", "Unt", "Unt", "H2O2", "H2O2", "Unt", "Unt", "H2O2", "H2O2",
"Unt", "Unt", "H2O2", "H2O2")
KD <- c("WT", "WT", "WT", "WT", "MLL1", "MLL1", "MLL1", "MLL1", "UTX", "UTX", "UTX", "UTX", "NRF2", "NRF2",
"NRF2", "NRF2")
Group <- c("CU", "CU", "CH", "CH", "MU", "MU", "MH", "MH", "UU", "UU", "UH", "UH", "NU", "NU", "NH", "NH")
sampleInfo <- data.frame(File, Rep, KD, Condition, Group)
sampleInfo # Check metadata
- Output:
> sampleInfo File Rep KD Condition Group 1 shControl_Unt_rep1 rep1 WT Unt CU 2 shControl_Unt_rep2 rep2 WT Unt CU 3 shControl_H2O2_rep1 rep1 WT H2O2 CH 4 shControl_H2O2_rep2 rep2 WT H2O2 CH 5 shMLL1_Unt_rep1 rep1 MLL1 Unt MU 6 shMLL1_Unt_rep2 rep2 MLL1 Unt MU 7 shMLL1_H2O2_rep1 rep1 MLL1 H2O2 MH 8 shMLL1_H2O2_rep2 rep2 MLL1 H2O2 MH 9 shUTX_Unt_rep1 rep1 UTX Unt UU 10 shUTX_Unt_rep2 rep2 UTX Unt UU 11 shUTX_H2O2_rep1 rep1 UTX H2O2 UH 12 shUTX_H2O2_rep2 rep2 UTX H2O2 UH 13 shNRF2_Unt_rep1 rep1 NRF2 Unt NU 14 shNRF2_Unt_rep2 rep2 NRF2 Unt NU 15 shNRF2_H2O2_rep1 rep1 NRF2 H2O2 NH 16 shNRF2_H2O2_rep2 rep2 NRF2 H2O2 NH
3. Load raw count table
- This example is based on the raw count table obtained via htseq-count.
- The raw count table is a tab-delimited
.txtfile where the first column contains gene or transcript IDs, and the remaining columns are the names of each sample. However, it lacks a header, making columns difficult to distinguish, and the last four rows contain summary information. - While you can open this file in Excel to add a header and remove the last 4 lines, I will perform these steps programmatically in R.
rawCountTable <- read.delim("~/Desktop/H2O2.txt", header = FALSE, row.names = NULL) rawCountTable <- rawCountTable[1:(nrow(rawCountTable) - 4), ] rownames(rawCountTable) <- rawCountTable[,1] rawCountTable <- rawCountTable[,-1] colnames(rawCountTable) <- c("shControl_Unt_rep1", "shControl_Unt_rep2", "shControl_H2O2_rep1", "shControl_H2O2_rep2", "shMLL1_Unt_rep1", "shMLL1_Unt_rep2", "shMLL1_H2O2_rep1", "shMLL1_H2O2_rep2", "shUTX_Unt_rep1", "shUTX_Unt_rep2", "shUTX_H2O2_rep1", "shUTX_H2O2_rep2", "shNRF2_Unt_rep1", "shNRF2_Unt_rep2", "shNRF2_H2O2_rep1", "shNRF2_H2O2_rep2") head(rawCountTable) # Check raw count table # After processing the data in Excel, you can load it in R using the following command: rawCountTable <- read.csv("~/Desktop/H2O2.csv", header = TRUE, row.names = 1) rawCountTable <- read.table("/path/to/the/file.txt", sep = ",", header = TRUE, row.names = 1)
- Output:
> head(rawCountTable) shControl_Unt_rep1 shControl_Unt_rep2 shControl_H2O2_rep1 shControl_H2O2_rep2 shMLL1_Unt_rep1 ENSG00000000003 0 12 1 1 1 ENSG00000000005 119 140 133 160 141 ENSG00000000419 1 1 1 1 1 ENSG00000000457 2 5 3 5 1 ENSG00000000460 273 196 279 282 242 ENSG00000000938 55 69 98 117 76 shMLL1_Unt_rep2 shMLL1_H2O2_rep1 shMLL1_H2O2_rep2 shUTX_Unt_rep1 shUTX_Unt_rep2 shUTX_H2O2_rep1 ENSG00000000003 1 1 1 0 1 0 ENSG00000000005 182 151 166 142 109 118 ENSG00000000419 2 2 2 2 3 1 ENSG00000000457 1 1 1 2 3 7 ENSG00000000460 193 218 209 214 235 203 ENSG00000000938 57 61 77 128 117 97 shUTX_H2O2_rep2 shNRF2_Unt_rep1 shNRF2_Unt_rep2 shNRF2_H2O2_rep1 shNRF2_H2O2_rep2 ENSG00000000003 1 1 2 1 0 ENSG00000000005 129 133 147 178 170 ENSG00000000419 0 1 1 0 0 ENSG00000000457 0 12 9 13 4 ENSG00000000460 204 100 171 179 180 ENSG00000000938 108 61 52 70 61
4. Object setup for RNA-seq analysis
- In this step, I will set up a
DGEListobject for edgeR analysis using the previously prepared raw count data and metadata.dgeFull <- DGEList(rawCountTable, group = sampleInfo$Group) dgeFull # check the objectKey arguments
rawCountTable: Specifies the object for raw count table.group = <Name>: Specifies the grouping of samples based on metadata, such as experimental conditions or sample types.
This grouping is crucial for DEG analysis in edgeR.
Output:
> dgeFull An object of class "DGEList" $counts shControl_Unt_rep1 shControl_Unt_rep2 shControl_H2O2_rep1 shControl_H2O2_rep2 shMLL1_Unt_rep1 ENSG00000000003 0 12 1 1 1 ENSG00000000005 119 140 133 160 141 ENSG00000000419 1 1 1 1 1 ENSG00000000457 2 5 3 5 1 ENSG00000000460 273 196 279 282 242 shMLL1_Unt_rep2 shMLL1_H2O2_rep1 shMLL1_H2O2_rep2 shUTX_Unt_rep1 shUTX_Unt_rep2 shUTX_H2O2_rep1 ENSG00000000003 1 1 1 0 1 0 ENSG00000000005 182 151 166 142 109 118 ENSG00000000419 2 2 2 2 3 1 ENSG00000000457 1 1 1 2 3 7 ENSG00000000460 193 218 209 214 235 203 shUTX_H2O2_rep2 shNRF2_Unt_rep1 shNRF2_Unt_rep2 shNRF2_H2O2_rep1 shNRF2_H2O2_rep2 ENSG00000000003 1 1 2 1 0 ENSG00000000005 129 133 147 178 170 ENSG00000000419 0 1 1 0 0 ENSG00000000457 0 12 9 13 4 ENSG00000000460 204 100 171 179 180 61494 more rows ... $samples group lib.size norm.factors shControl_Unt_rep1 CU 11738068 1 shControl_Unt_rep2 CU 11905476 1 shControl_H2O2_rep1 CH 12069056 1 shControl_H2O2_rep2 CH 12237122 1 shMLL1_Unt_rep1 MU 11781709 1 11 more rows ...
5. Check Library Size and Distribution
- In this step, I will check the library size and distribution of the samples before proceeding with normalization.
pseudoCounts <- log2(dgeFull$counts+1) # the values are converted to non-zero values. FullCol <- as.numeric(factor(sampleInfo$Group)) + 1 boxplot(pseudoCounts, col=FullCol, las=2) abline(h=median(as.matrix(pseudoCounts)), col="blue") # Save boxplotOutput:
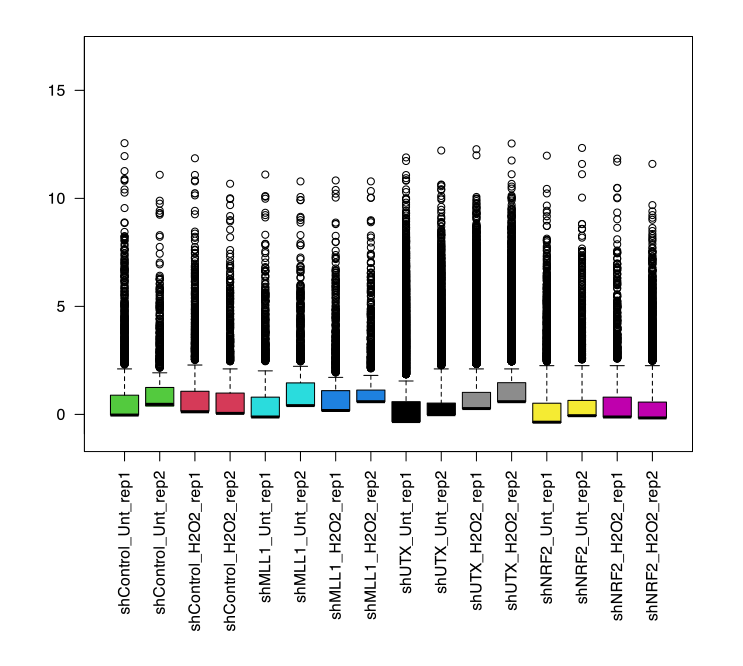
6. TMM Normalization
- In this process, I will perform TMM normalization.
dgeFull <- DGEList(dgeFull$counts[apply(dgeFull$counts, 1, sum) != 0, ], group=dgeFull$samples$group) dge <- calcNormFactors(dgeFull) design <- model.matrix(~0+group, data=dge$samples) dge <- estimateDisp(dge, design = design) - in these codes,
- Line 1: Creates a DGEList object with count data, excluding genes with zero counts across all samples, and includes group information. This step improves data quality by removing noise.
- Line 2: Calculates TMM normalization factors to adjust for differences in library sizes between samples, ensuring accurate comparisons.
- Line 3: Constructs a design matrix for DEG analysis, indicating group membership of each sample. The
~0+groupcommand facilitates group comparisons. - Line 4: Estimates the dispersion for each gene, crucial for reliable DEG analysis, by accounting for variability in gene expression levels.
Output:
> dgeFull <- DGEList(dgeFull$counts[apply(dgeFull$counts, 1, sum) != 0, ], group=dgeFull$samples$group) > dgeFull$counts # **Check the selected transcripts. I will not show results in this example because it is too long. > dge <- calcNormFactors(dgeFull) > dge$samples # Check norm.factor group lib.size norm.factors shControl_Unt_rep1 CU 11738068 1.0531166 shControl_Unt_rep2 CU 11905476 0.8733873 shControl_H2O2_rep1 CH 12069056 1.1370291 shControl_H2O2_rep2 CH 12237122 0.8497171 shMLL1_Unt_rep1 MU 11781709 1.1008688 shMLL1_Unt_rep2 MU 11949117 0.8701975 shMLL1_H2O2_rep1 MH 10394452 1.0873398 shMLL1_H2O2_rep2 MH 10562518 0.9844330 shUTX_Unt_rep1 UU 11606646 1.1195918 shUTX_Unt_rep2 UU 11774054 0.8831360 shUTX_H2O2_rep1 UH 10663731 1.1049622 shUTX_H2O2_rep2 UH 10831797 0.9599600 shNRF2_Unt_rep1 NU 10051201 1.0067264 shNRF2_Unt_rep2 NU 10218609 1.0175643 shNRF2_H2O2_rep1 NH 10533324 1.0512145 shNRF2_H2O2_rep2 NH 10701390 0.9716580After caculation of norm.factors:
> design <- model.matrix(~0+group, data=dge$samples) > design groupCH groupCU groupMH groupMU groupNH groupNU groupUH groupUU shControl_Unt_rep1 0 1 0 0 0 0 0 0 shControl_Unt_rep2 0 1 0 0 0 0 0 0 shControl_H2O2_rep1 1 0 0 0 0 0 0 0 shControl_H2O2_rep2 1 0 0 0 0 0 0 0 shMLL1_Unt_rep1 0 0 0 1 0 0 0 0 shMLL1_Unt_rep2 0 0 0 1 0 0 0 0 shMLL1_H2O2_rep1 0 0 1 0 0 0 0 0 shMLL1_H2O2_rep2 0 0 1 0 0 0 0 0 shUTX_Unt_rep1 0 0 0 0 0 0 0 1 shUTX_Unt_rep2 0 0 0 0 0 0 0 1 shUTX_H2O2_rep1 0 0 0 0 0 0 1 0 shUTX_H2O2_rep2 0 0 0 0 0 0 1 0 shNRF2_Unt_rep1 0 0 0 0 0 1 0 0 shNRF2_Unt_rep2 0 0 0 0 0 1 0 0 shNRF2_H2O2_rep1 0 0 0 0 1 0 0 0 shNRF2_H2O2_rep2 0 0 0 0 1 0 0 0 attr(,"assign") [1] 1 1 1 1 1 1 1 1 attr(,"contrasts") attr(,"contrasts")$group [1] "contr.treatment"
7. Check Library Size and Distribution after Normalization
- Next, I will now plot a boxplot based on the normalized data to observe the correlation between each sample or between groups.
- Depicting a box plot
normCounts <- cpm(dge) pseudoNormCounts <- log2(normCounts + 1) FullColNorm <- as.numeric(factor(dge$samples$group)) + 1 boxplot(pseudoNormCounts, col=FullColNorm, las=2) abline(h=median(as.matrix(pseudoNormCounts)), col="blue") # can save a plot - Output:
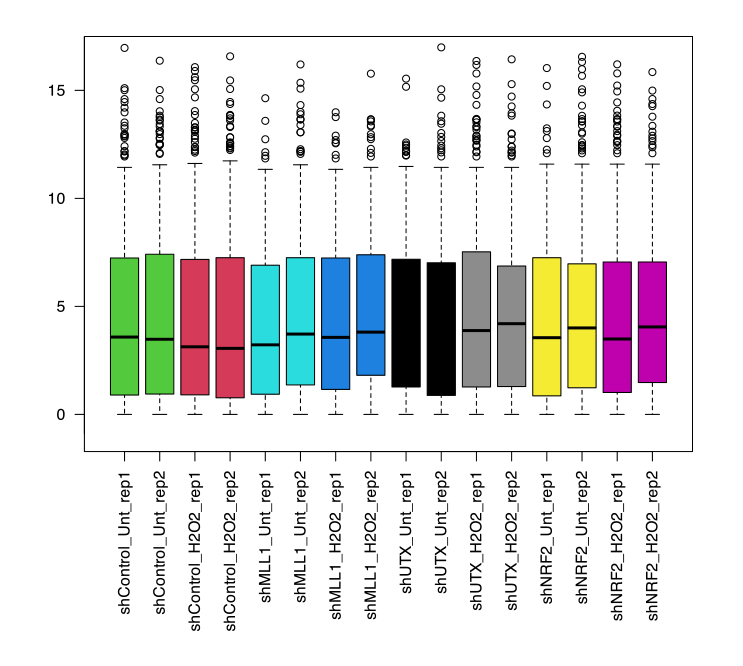
- Drwing a principal component analysis (PCA) plot: PCA visualizes the major sources of variation in the data by transforming it into principal components. It highlights the directions (principal components) where the data varies the most, helping to identify patterns and groupings among the samples. The PCA plot is useful for understanding the overall structure and key sources of variation in the dataset.
pca_result <- prcomp(t(pseudoNormCounts), scale. = TRUE) summary(pca_result) # PCA Visulaize pca_data <- data.frame(Sample = rownames(pca_result$x), PC1 = pca_result$x[,1], PC2 = pca_result$x[,2], Group = dge$samples$group) # PCA plot ggplot(pca_data, aes(x = PC1, y = PC2, color = Group)) + geom_point(size = 3) + labs(title = "PCA Plot", x = "Principal Component 1", y = "Principal Component 2") + theme_minimal() - Making a multidimensional scaling (MDS) plot: MDS plots represent the pairwise distances between samples in a low-dimensional space, preserving the original distances as much as possible. This helps in visualizing the similarity or dissimilarity among samples based on their expression profiles. The MDS plot is particularly useful for detecting patterns and relationships in the data that are not immediately apparent.
# Basal format plotMDS(pseudoNormCounts) # Advanced format plotMDS(pseudoNormCounts, col = FullColNorm, pch = 19, main = "MDS Plot") legend("topright", legend=levels(factor(dge$samples$group)), col = 1:length(levels(factor(dge$samples$group))), pch = 19) - Prepare log10 transformed CPM
prior.count = 1 cpmlog <- cpm(dge, log = T, prior.count = prior.count) write.csv(cpmlog, "path/to/the/file.csv") # Save the CPM file
8. Remove Overdispersion and DEG Analysis
In this step, I will perform DEG analysis based on the TMM normalized counts (
dge) and thedesignmatrix. Overdispersion, which is the presence of greater variability in the data than expected under a negative binomial or Poisson distribution, must be addressed to ensure accurate results.This can be effectively removed using linear regression approaches, such as the Generalized Linear Model-Quasi Likelihood (GLM-QL) method. The GLM-QL approach accounts for overdispersion by modeling the extra variability and provides more reliable statistical inferences.
If no specific parameters are added, the GLM-QL typically calculates statistical significance by comparing the degrees of freedom between samples. Additionally, the false discovery rate (FDR) is computed using the Benjamini-Hochberg (BH) method to control for multiple testing and reduce the likelihood of false positives.
# Model fitting using the GML-QL and modeling overdispersion fit <- glmQLFit(dge, design = design) glmQLFTest(fit) # (Optional) Test QL F-test across all samples # The process of specifying what to compare based on design object my.contrasts <- makeContrasts(CU_CH = groupCH-groupCU, CU_MU = groupMU-groupCU, CU_UU = groupUU-groupCU, CU_NU = groupNU-groupCU, MU_MH = groupMH-groupMU, UU_UH = groupUH-groupUU, NU_NH = groupNH-groupNU, CH_MH = groupMH-groupCH, CH_UH = groupUH-groupCH, CH_NH = groupNH-groupCH, levels = design) # DEG Analysis H2O2 <- glmQLFTest(fit, contrast=my.contrasts[,"CU_CH"]) # DEG analysis using the QL F-test under the [my.contrasts] conditions. H2O2_FDR <- topTags(H2O2, n = Inf)$table # FDR calculation with BH (default) method write.csv(H2O2_FDR, "H2O2_DEG.csv") # Save as csv format shMLL1 <- glmQLFTest(fit, contrast=my.contrasts[,"MU_MH"]) shMLL1_FDR <- topTags(shMLL1, n = Inf)$table write.csv(shMLL1_FDR, "shMLL1_DEG.csv") shUTX <- glmQLFTest(fit, contrast=my.contrasts[,"UU_UH"]) shUTX_FDR <- topTags(shUTX, n = Inf)$table write.csv(shUTX_FDR, "shUTX_DEG.csv") shNRF2 <- glmQLFTest(fit, contrast=my.contrasts[,"NU_NH"]) shNRF2_FDR <- topTags(shNRF2, n = Inf)$table write.csv(shNRF2_FDR, "shNRF2_DEG.csv")
- Output:
logFCrepresents the log2-transformed value of the target group’s (H2O2) expression divided by the control group’s (Unt) expression, whilelogCPMis the average of log2(CPM + 1) for each sample. TheFvalue is the result of the F-test (glmQL), and thePValue(p-value) indicates the statistical significance of the result.FDRis the False Discovery Rate, which is a multiple testing correction of the p-value. In general, values with $|logFC|$ > 1 and FDR < 0.05 are considered significant DEGs (see the bellow).> head(H2O2_FDR) logFC logCPM F PValue FDR ENSG00000261089 -7.753531 3.5081044 1499.6617 0.000000e+00 0.000000e+00 ENSG00000205358 -8.403098 1.3033323 706.6869 1.355224e-155 4.167246e-151 ENSG00000181019 3.767159 2.5203353 486.9699 7.338978e-108 1.504466e-103 ENSG00000181625 -5.324815 1.1710369 311.2585 1.220608e-69 1.876655e-65 ENSG00000204472 6.711876 0.8264076 227.5472 2.097907e-51 2.580384e-47 ENSG00000155070 -1.582992 3.5280535 148.1809 4.380133e-34 4.489563e-30
- More genes with FDR > 0.05 could emerge in your actual DEG analysis, despite the distinctiveness evident in the PCA and MDS plot between the samples from the two conditions under comparison.
- In this case, it may be helpful to change the test for statistical significance to fitness (glmLRT; Generalized Linear Model Likelihood Ratio Test function) rather than degrees of freedom (glmQL function).
Here is an example of R code for DEG analysis using the glmLRT:
fit <- glmQLFit(dge, design = design) # DEG analysis using the Likelihood Ratio Test H2O2_LRT <- glmLRT(fit, contrast=my.contrasts[,"CU_CH"]) H2O2_LRT_FDR <- topTags(H2O2_LRT, n=Inf)$table # p.adjust.method = "[BY/holm/etc]" write.csv(H2O2_LRT_FDR, "H2O2_DEG_glmLRT.csv")- There are several methods to determine the FDR value based on statistical analysis, including
BH,BY,holm,bonferroni,hommel,hochberg, ornone, but theBH(default) method has greater tolerance rather than other methods and most users acceptBH. You can choose these methods from thetopTagscommand as shown below:shMLL1_FDR <- topTags(shMLL1, n=Inf, p.adjust.method = "<BY/holm/etc>")$table
9. Single Sample DEG analysis: Importance of Biological Replicates in RNA-seq
Biological replicates in RNA-seq are crucial for ensuring reliable, reproducible results by accounting for natural biological variability. They improve statistical power, ensure reproducibility, and reduce bias. However, researchers may be constrained to single samples due to economic limitations, scarcity of samples, or ethical considerations. Despite these constraints, single-sample RNA-seq can still provide valuable insights, though results must be interpreted with caution, acknowledging the limitations imposed by the lack of biological replicates.
In particular, the coefficient of dispersion between samples cannot be calculated directly, necessitating the addition of an arbitrary factor. This factor is referred to as the biological coefficient of variation (BCV). Researchers can estimate this value based on prior knowledge or data from analogous experiments.
It is crucial to acknowledge that the inability to calculate the dispersion coefficient implies that linear models and methods for removing overdispersion will not be effective.
Typically, a BCV value of
0.4is utilized. If the dataset under analysis is anticipated to exhibit high biological variation, the BCV value can be set to0.6or higher. For instance, samples from different species or those collected under varying environmental conditions might necessitate a higher BCV value. Conversely, for datasets with low biological variation, a BCV value of0.2may be appropriate. For example, a low BCV value might be used when repeatedly collecting the same cell line under identical conditions. These values should be considered as guidelines rather than strict rules.There are two methods to apply BCV to perform DEG analysis, the
extractTestandglmFitmethods. Until the normalization step, both methods are same. Here, I will provide how to use both methods after normalization:extractTestmethod:dge <- calcNormFactors(dgeFull) # TMM normalization bcv <- 0.4 dgeTest_H2O2 <- exactTest(dgeFull, dispersion = bcv^2, pair = c("CU","CH")) dgeTest_H2O2_FDR <- topTags(dgeTest_H2O2, n = Inf)$table write.csv(dgeTest_H2O2_FDR, "H2O2_DEG_extract,csv")Output
> dgeTest_H2O2 <- exactTest(dgeFull, dispersion = bcv^2, pair = c("CU","CH")) > dgeTest_H2O2_FDR <- topTags(dgeTest_H2O2, n = Inf)$table > dgeTest_H2O2_FDR <- dgeTest_H2O2_FDR[order(rownames(dgeTest_H2O2_FDR)), ] > head(dgeTest_H2O2_FDR) logFC logCPM PValue FDR ENSG00000000003 0.00000000 -1.5072179 1.0000000 1 ENSG00000000005 0.01620179 4.6487540 1.0000000 1 ENSG00000000419 -3.25162822 -1.0936018 1.0000000 1 ENSG00000000457 0.78712182 -0.4296291 0.7447837 1 ENSG00000000460 -0.14629616 5.1399138 0.8668006 1 ENSG00000000938 0.71112235 3.7377811 0.4122082 1glmFitmethods:dge <- calcNormFactors(dgeFull) # TMM normalization design <- model.matrix(~0+group, data=dge$samples) # design matrix bcv <- 0.4 my.contrasts <- makeContrasts(CU_CH = groupCH-groupCU, CU_MU = groupMU-groupCU, CU_UU = groupUU-groupCU, CU_NU = groupNU-groupCU, MU_MH = groupMH-groupMU, UU_UH = groupUH-groupUU, NU_NH = groupNH-groupNU, CH_MH = groupMH-groupCH, CH_UH = groupUH-groupCH, CH_NH = groupNH-groupCH, levels = design) fit <- glmFit(dge, design = design, dispersion=bcv^2) glmLRT(fit) IND_H2O2_LRT <- glmLRT(fit, contrast=my.contrasts[,"CU_CH"]) IND_H2O2_LRT_FDR <- topTags(IND_H2O2_LRT, n=Inf)$table write.csv(IND_H2O2_LRT_FDR, "H2O2_DEG_LRT.csv")Output
> IND_H2O2_LRT <- glmLRT(fit, contrast=my.contrasts[,"CU_CH"]) > IND_H2O2_LRT_FDR <- topTags(IND_H2O2_LRT, n=Inf)$table > IND_H2O2_LRT_FDR <- IND_H2O2_LRT_FDR[order(rownames(IND_H2O2_LRT_FDR)), ] > head(IND_H2O2_LRT_FDR) logFC logCPM LR PValue FDR ENSG00000000003 0.0000000 -1.5072179 0.0000000000 1.0000000 1.0000000 ENSG00000000005 0.0162005 4.6487540 0.0003788167 0.9844716 1.0000000 ENSG00000000419 -3.1350554 -1.0936018 1.4297184098 0.2318104 0.9934706 ENSG00000000457 0.7839430 -0.4296291 0.3116571808 0.5766650 1.0000000 ENSG00000000460 -0.1462899 5.1399138 0.0314383257 0.8592660 1.0000000 ENSG00000000938 0.7110305 3.7377811 0.7053500976 0.4009918 1.0000000
10. Annotation
After performing DEG analysis, the results may be displayed using gene (or transcript) IDs instead of gene symbols. To convert these IDs into various information such as gene symbols and biotypes, annotation is required. Two main methods for this are using the biomaRt R package or extracting information directly from a GTF file.
Biomart (
biomaRt) offers access to a vast array of biological data through the Ensembl database, facilitating the easy conversion of gene IDs to gene symbols. However, this conversion can be more challenging and complex for species other than humans and mice. Therefore, it is recommended to use Biomart for gene conversion in the cases of human and mouse data.Alternatively, the GTF file method entails extracting information from a locally stored gene annotation file. By parsing the GTF file, one can map gene IDs to gene symbols. This approach is versatile and can be applied to any species, provided that a GTF file is available.
the biomaRt method for human and mouse data:
mart <- useDataset("hsapiens_gene_ensembl", useMart("ensembl")) anno <- getBM(filters = "ensembl_gene_id", attributes= c("ensembl_gene_id", "gene_biotype", "external_gene_name"), values = row.names(IND_H2O2_LRT_FDR), mart = mart) Compare <- setDT(IND_H2O2_LRT_FDR, keep.rownames = "ensembl_gene_id")[] MergeDEG <- merge(IND_H2O2_LRT_FDR, anno, by="ensembl_gene_id", all=TRUE) write.csv(MergeDEG, "IND_H2O2_LRT_Annotated.csv")- in these codes,
- Line 1: Connects to the Ensembl database (
useMart("ensembl")) and specifies the dataset to use (useDataset), in this case, the human gene dataset from Ensembl (mouse:"mmusculus_gene_ensembl"). The availableuseDatasetcan be found via thelistDatasets(mart)command, note the case of the lists. - Line 2:
filtersspecifies that the filter for the query is the Ensembl gene ID.attributesdefines the attributes to retrieve, including the Ensembl gene ID, gene biotype, and external gene name (gene symbol).valuesprovides the list of Ensembl gene IDs to be annotated, which are the row names of theIND_H2O2_LRT_FDRobject. The availableattributescan be found via thelistAttributes(mart)command, note the case of the lists. - Line 3: Converts
IND_H2O2_LRT_FDRto a data.table and keeps the row names as a column namedensembl_gene_id.[]ensures the operation is executed immediately and the result is assigned to Compare. - Line 4: Merges the DEG results (
IND_H2O2_LRT_FDR) with the annotation information (anno) based on theby = "ensembl_gene_id"column. Theall = TRUEargument ensures that all entries from both data frames are included in the result, performing a full outer join.
Output
> mart <- useDataset("hsapiens_gene_ensembl", useMart("ensembl")) > anno <- getBM(filters= "ensembl_gene_id", attributes= c("ensembl_gene_id", "gene_biotype", "external_gene_name"), values = row.names(IND_H2O2_LRT_FDR),mart= mart) > head(anno) ensembl_gene_id gene_biotype external_gene_name 1 ENSG00000000003 protein_coding TSPAN6 2 ENSG00000000005 protein_coding TNMD 3 ENSG00000000419 protein_coding DPM1 4 ENSG00000000457 protein_coding SCYL3 5 ENSG00000000460 protein_coding FIRRM 6 ENSG00000000938 protein_coding FGR > Compare <- setDT(IND_H2O2_LRT_FDR, keep.rownames = "ensembl_gene_id")[] > head(Compare) ensembl_gene_id logFC logCPM LR PValue FDR <char> <num> <num> <num> <num> <num> 1: ENSG00000000003 0.0000000 -1.5072179 0.0000000000 1.0000000 1.0000000 2: ENSG00000000005 0.0162005 4.6487540 0.0003788167 0.9844716 1.0000000 3: ENSG00000000419 -3.1350554 -1.0936018 1.4297184098 0.2318104 0.9934706 4: ENSG00000000457 0.7839430 -0.4296291 0.3116571808 0.5766650 1.0000000 5: ENSG00000000460 -0.1462899 5.1399138 0.0314383257 0.8592660 1.0000000 6: ENSG00000000938 0.7110305 3.7377811 0.7053500976 0.4009918 1.0000000 > MergeDEG <- merge(IND_H2O2_LRT_FDR, anno, by="ensembl_gene_id", all=TRUE) > head(MergeDEG) Key: <ensembl_gene_id> ensembl_gene_id logFC logCPM LR PValue FDR gene_biotype external_gene_name <char> <num> <num> <num> <num> <num> <char> <char> 1: ENSG00000000003 0.0000000 -1.5072179 0.0000000000 1.0000000 1.0000000 protein_coding TSPAN6 2: ENSG00000000005 0.0162005 4.6487540 0.0003788167 0.9844716 1.0000000 protein_coding TNMD 3: ENSG00000000419 -3.1350554 -1.0936018 1.4297184098 0.2318104 0.9934706 protein_coding DPM1 4: ENSG00000000457 0.7839430 -0.4296291 0.3116571808 0.5766650 1.0000000 protein_coding SCYL3 5: ENSG00000000460 -0.1462899 5.1399138 0.0314383257 0.8592660 1.0000000 protein_coding FIRRM 6: ENSG00000000938 0.7110305 3.7377811 0.7053500976 0.4009918 1.0000000 protein_coding FGR - Line 1: Connects to the Ensembl database (
- in these codes,
- the GTF file method for zebrafish:
gtf_path <- "/Users/jchoi/Desktop/Danio_rerio.GRCz11.111.gtf" gtf_data <- rtracklayer::import(gtf_path) gene_info <- subset(gtf_data, type == "gene") all_attributes <- elementMetadata(gene_info) gene_attributes_df <- data.frame(all_attributes, stringsAsFactors = FALSE) write.csv(gene_attributes_df, "annot.csv")- in these codes,
- Line 1: Specify the GTF file path.
- Line 2: Import the information from the GTF file using
rtracklayerpackage. - Line 3: Creates a subset of
gtf_datathat includes only the entries where the type is"gene". Thegene_infoobject will now contain only the gene-level annotations. - Line 4: Extracts the metadata (
attributes) of thegene_infoobject using theelementMetadatafunction. Theall_attributesobject will contain all theattributesof the gene annotations. - Line 5: Converts the
all_attributesobject into a data frame namedgene_attributes_dfusing thedata.framefunction. ThestringsAsFactors = FALSEargument ensures that string columns are not converted to factors.
Output
> gene_attributes_df <- data.frame(all_attributes, stringsAsFactors = FALSE) > head(gene_attributes_df) source type score phase gene_id gene_version gene_name gene_source gene_biotype 1 havana gene NA NA ENSDARG00000103202 2 CR383668.1 havana lincRNA 2 ensembl_havana gene NA NA ENSDARG00000009657 8 fgfr1op2 ensembl_havana protein_coding 3 havana gene NA NA ENSDARG00000096472 2 AL845295.2 havana processed_transcript 4 havana gene NA NA ENSDARG00000096156 3 si:dkey-21h14.12 havana protein_coding 5 havana gene NA NA ENSDARG00000076160 6 si:dkey-285e18.2 havana protein_coding 6 havana gene NA NA ENSDARG00000117163 1 znf1114 havana protein_coding transcript_id transcript_version transcript_name transcript_source transcript_biotype tag exon_number exon_id 1 <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> 2 <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> 3 <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> 4 <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> 5 <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> 6 <NA> <NA> <NA> <NA> <NA> <NA> <NA> <NA> exon_version protein_id protein_version 1 <NA> <NA> <NA> 2 <NA> <NA> <NA> 3 <NA> <NA> <NA> 4 <NA> <NA> <NA> 5 <NA> <NA> <NA> 6 <NA> <NA> <NA>
- in these codes,
- For some other species (e.g. rice), there are also symbol conversion packages:
library(riceidconverter) library(org.Osativa.eg.db) RAP <- RiceIDConvert(myID = data$Name, fromType = "MSU", toType = "RAP") Symbol <- RiceIDConvert(myID = data$MSU, fromType = "MSU", toType = "SYMBOL") ## Symbol indicates Gene ID (LOCXXXXXXX format)
Citations
- Robinson, M. D., McCarthy, D. J., & Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. bioinformatics, 26(1), 139-140. DOI
- McCarthy, D. J., Chen, Y., & Smyth, G. K. (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic acids research, 40(10), 4288-4297. DOI
- Ritchie, M. E., Phipson, B., Wu, D. I., Hu, Y., Law, C. W., Shi, W., & Smyth, G. K. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research, 43(7), e47-e47. DOI